Retinoblastome
Retinoblastome: Definition, Entstehung und Häufigkeit
Was sind Retinoblastome?
Das Retinoblastom ist ein bösartiger (maligner) Tumor der Netzhaut (Retina), der vor allem bei sehr jungen Kindern – Säuglingen und Kleinkindern vor dem fünften Lebensjahr – auftritt. Obwohl die Erkrankung insgesamt eher selten ist, zählen Retinoblastome bei Kindern zu den am häufigsten auftretenden kindlichen Augentumoren weltweit. Bei frühzeitiger Diagnose ist die Retinoblastom Lebenserwartung vielversprechend – mehr als 95 Prozent der Patientinnen und Patienten überleben einen Netzhauttumor.
Slider: Darstellung gesundes Auge (Slide 1) versus Auge mit Retinoblastom (Slide 2).
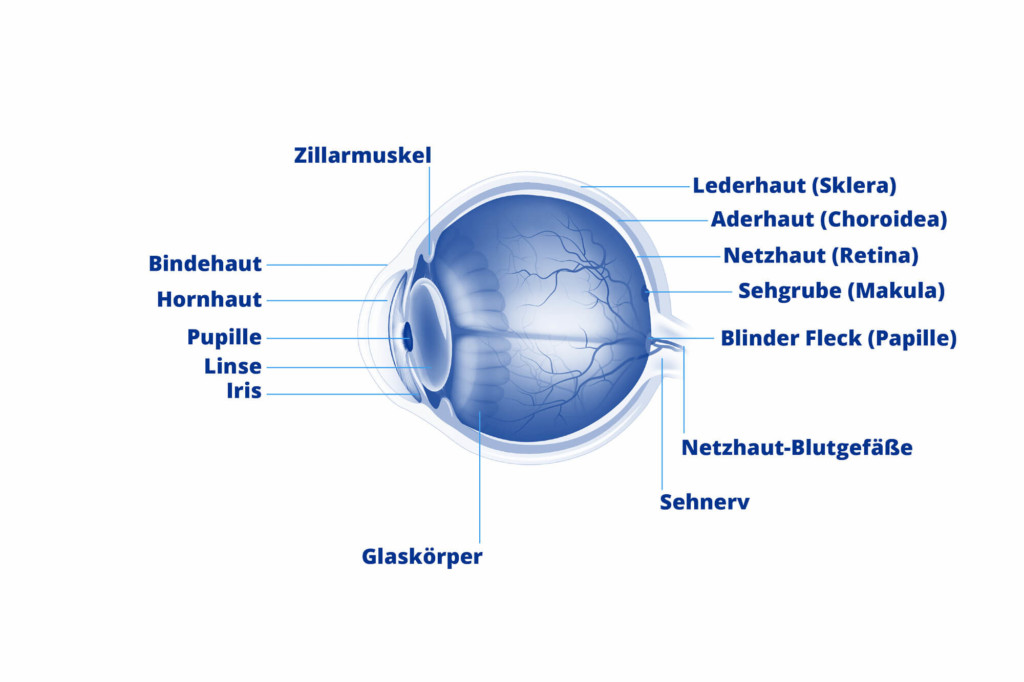
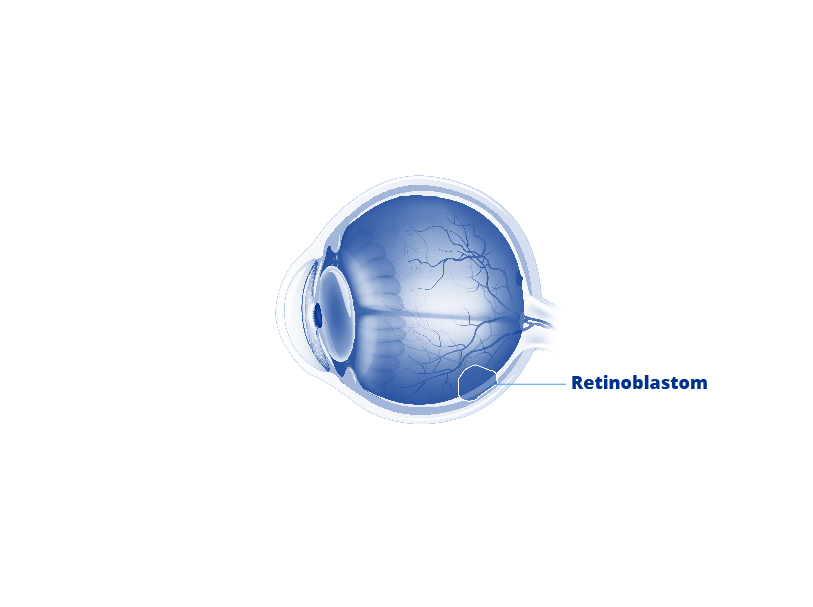
Retinoblastom Ursachen
Netzhauttumoren entwickeln sich aus noch unreifen Zellen der Retina. Die Ursache für die Entartung dieser Zellen ist genetisch bedingt: Die Veränderungen betreffen dabei das so genannte Retinoblastom-Gen (RB1-Gen). Entweder findet sich die entsprechende Mutation in den entarteten unreifen Netzhautzellen selbst oder sie ist grundsätzlich in allen Keimzellen des Körpers verankert. Im letzteren Fall entsteht das Retinoblastom durch Vererbung. Mehrheitlich entstehen die Mutationen jedoch neu (sporadisch) und können nur in den Tumorzellen nachgewiesen werden.
Welche Arten von Retinoblastomen gibt es?
etinoblastome lassen sich anhand verschiedener Kriterien unterscheiden:
Nach Lokalisation des Tumors:
- einseitiges (unilaterales) Retinoblastom – betroffen ist nur ein Auge (etwa zwei Drittel aller Fälle),
- beidseitiges (bilaterales) Retinoblastom – betroffen sind beide Augen.
Nach Anzahl der Tumoren bzw. konkreter Lokalisation innerhalb des Auges:
- unifokal – betroffen ist nur eine Stelle im Auge,
- multifokal – es finden sich an mehreren Stellen im Auge Tumoren.
Unsere Experten
Klinikdirektorin
Univ.-Prof. Dr. Beate Timmermann
Spezialist:in im OnkoZert-Behandlungsprogram Augentumoren

Dr. med.
Ronald Richter
Oberarzt,
Facharzt für Strahlentherapie
Kontakt
Sie möchten sich über die Protonentherapie am WPE informieren? Unser Case Management ist für Sie da. Dieses klärt sämtliche Anfragen gemeinsam mit Ihnen & unseren Spezialist:innen.
Telefon: 0201 723 6600
Publikation
Prof. Beate Timmermann hat in der neuen Auflage des Standardwerks für Radioonkologie Perez and Brady’s Principles and Practice of Radiation Oncology das Kapitel zum Retinoblastom verfasst. Gemeinsam mit John T. Lucas Jr. stellt sie darin den aktuellen Stand der Therapie und den gezielten Einsatz der Strahlentherapie dar.
Nach Fortschritt des Tumorwachstums:
- intraokulares Retinoblastom – befallen ist/sind nur unmittelbar das Auge/die Augen,
- extraokulares Retinoblastom – der Tumor ist bereits in umliegende Areale hineingewachsen.
Nach genetischer Besonderheit:
- erbliche Retinoblastome – die Mutation findet sich in allen Keimzellen des Körpers (germinale Mutation), mehrheitlich beidseitig, häufig multifokal sowie
- nicht-erbliche Retinoblastome – die Mutation findet sich nur in den Tumorzellen und entsteht neu (sporadisch) und zwar immer einseitig und in der Regel unifokal (in etwa 60 Prozent aller Fälle).
Sind bei einem erblichen Retinoblastom bereits Erkrankungen innerhalb der Familie bekannt, kann man davon ausgehen, dass das veränderte Gen von einem Elternteil an das Kind weitergegeben wurde (familiäres Retinoblastom). Laut Angaben der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) ist dies bei etwa einem Viertel der betroffenen Patientinnen und Patienten mit erblichem Netzhauttumor der Fall. Bei sporadisch entstandenem erblichen Retinoblastomen hat sich die Mutation dagegen erst neu in den Keimzellen des Körpers entwickelt. Kinder, die die Genmutation geerbt haben, erkranken laut GOPH beinahe immer an einem Retinoblastom (beinahe zu 100 Prozent) und können die Erkrankung auch selbst an mögliche Nachkommen weitergeben. Zudem besteht die Gefahr, dass auch Geschwisterkinder erkrankt sind oder sich andere Krebserkrankungen entwickeln. Familiäre Retinoblastome gehören damit zu den so genannten Krebsprädispositionssyndromen.
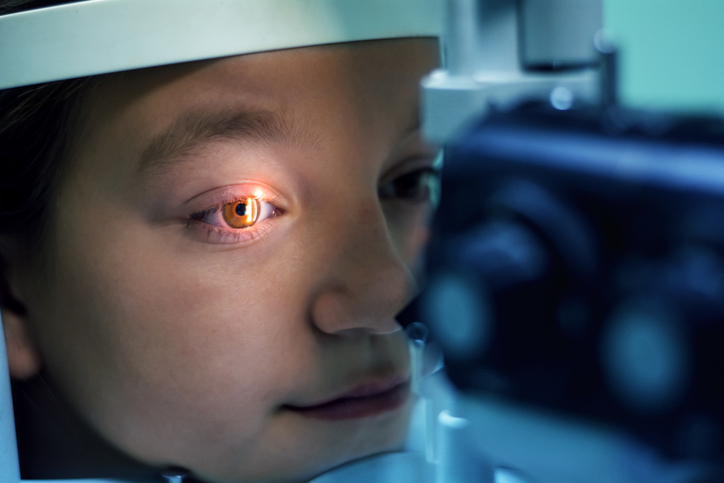
Seltene Sonderformen: Trilaterales Retinoblastom & Retinom
In Einzelfällen können sehr seltene Sonderformen eines Retinoblastoms auftreten. Beim so genannten trilateralen Retinoblastom liegen gleichzeitig ein erbliches Retinoblastom sowie ein eigenständig wachsender Hirntumor im Bereich der Zirbeldrüse (Pinealoblastom) vor. Ebenso selten ist das Retinom – ein Retinoblastom, das sich von selbst wieder zurückgebildet hat. Man spricht hier auch von einer klinisch stummen Verlaufsform der Erkrankung.
Wie häufig sind Retinoblastome?
Retinoblastome gehören zu den seltenen Krebserkrankungen. In der Gruppe der malignen (bösartigen) Krebserkrankungen im Kindes- und Jugendalter kommen sie laut Angaben des Deutschen Kinderkrebsregisters auf einen Anteil von etwa zwei Prozent. Deutschlandweit erkranken pro Jahr etwa 40 bis 60 Kinder an einem Netzhauttumor. Da sich das Retinoblastom aus noch unreifen Zellen der Retina entwickelt, tritt der Tumor in aller Regel noch vor dem fünften Lebensjahr auf. Erkrankungen bei älteren Kindern und Erwachsenen sind möglich, aber noch seltener. Einseitige Retinoblastome werden vielfach bei Kindern bis zu zwei Jahren diagnostiziert. Das durchschnittliche Alter, zu dem die beidseitige Variante festgestellt wird, beträgt ein Jahr. Gut zehn Prozent aller Tumoren können bereits bei der Geburt diagnostiziert werden.
Wo entsteht ein Netzhauttumor, und kann er Metastasen bilden?
Retinoblastome gehören zu den schnell wachsenden Tumoren. Ausgehend von der Netzhaut können sie sich weiter im Augen- und Schädelbereich ausbreiten und beispielsweise in die Augenhöhle und letztlich über den Sehnerv bis in das Gehirn und das Zentralnervensystem vordringen. Auch ist eine Streuung (Metastasierung) über Blut und Lymphe in andere Organe möglich. Typisch ist zudem die Ausbildung gleich mehrerer Tumoren. Dies geschieht unter anderem, indem sich einzelne Tumorzellen von der Netzhaut lösen, im gelartigen Glaskörper des Auges „schweben“ und an anderer Stelle der Netzhaut wieder andocken (Glaskörperaussaat).
Welche Stadien gibt es bei einem Retinoblastom?
Um das genaue Stadium der Krebserkrankung zu bestimmen, nutzen Fachleute bei Retinoblastomen verschiedene Klassifikationssysteme. Anhand dieser Systeme wird der genaue Stand der Tumorausbreitung festgelegt – und zwar für jedes Auge einzeln. Die erste wichtige Frage lautet dabei: Ist der Tumor noch auf das Auge beschränkt (intraokular) oder bereits in andere Bereiche hineingewachsen (extraokular)?
Je nach Größe, Lokalisation und Anzahl der Tumoren wird bei intraokularen Retinoblastomen zwischen fünf Stadien (Stadium A bis E) unterschieden. Angefangen von einzelnen kleinen Tumoren, die auf die Netzhaut beschränkt bleiben und eine ausreichende Entfernung zum Sehnerv (mindestens 1,5 Millimeter) und anderen sehrelevanten Strukturen haben, bis hin zu großen, augenfüllenden Retinoblastomen, die bereits in andere Areale des Kopfes vorgedrungen sind. In diesen Fällen ist ein Erhalt des Auges nicht mehr möglich. Bei extraokularen Netzhauttumoren wird international zwischen vier Stadien differenziert, abhängig unter anderem davon, ob der Sehnerv befallen ist und ob das Blastom bereits gestreut hat oder nicht.
Die exakte Festlegung des Stadiums der Erkrankung ist nicht nur entscheidend für die Wahl der zur Verfügung stehenden Therapieoptionen, sondern gibt unter Umständen auch Aufschluss darüber, inwieweit die Sehfähigkeit erhalten werden kann bzw. ob das Auge an sich noch zu retten ist oder lieber chirurgisch entfernt werden sollte.
Welche Retinoblastom Symptome können auftreten?
Netzhauttumore bleiben oft lange Zeit unentdeckt, da sich das Auge zunächst äußerlich nicht verändert. Bei der einseitigen Variante wird zudem die Sehverschlechterung des betroffenen Auges durch das gesunde Auge zunächst kompensiert. Meist wird ein Blastom erst dann diagnostiziert, wenn der Tumor bereits eine gewisse Größe erreicht hat oder in andere Bereiche hineinwächst.
Eines der häufigsten Symptome für ein Retinoblastom ist eine so genannte Leukokorie („Katzenauge“), eine weiß leuchtende Pupille. Weitere mögliche Anzeichen für einen Netzhauttumor sind außerdem:
- ein Anstieg des Augeninnendrucks,
- ein schmerzendes Auge bzw. Augen- und Lidentzündungen,
- Farbveränderungen der Iris,
- ein sich verschlechterndes Sehvermögen,
- Schielfehlstellungen (Strabismus) sowie
- Pupillenveränderungen (beispielsweise weite Pupillen).
Um unter anderem ein Retinoblastom so früh wie möglich zu erkennen, ist mittlerweile der so genannte „Brückner-Test“ fester Bestandteil der Vorsorgeuntersuchungen (U-Untersuchungen) bei Kleinkindern. Bei diesem ophtalmologischen Verfahren (Ophtalmologie: Augenheilkunde) werden die Augen durchleuchtet, um Veränderungen festzustellen.
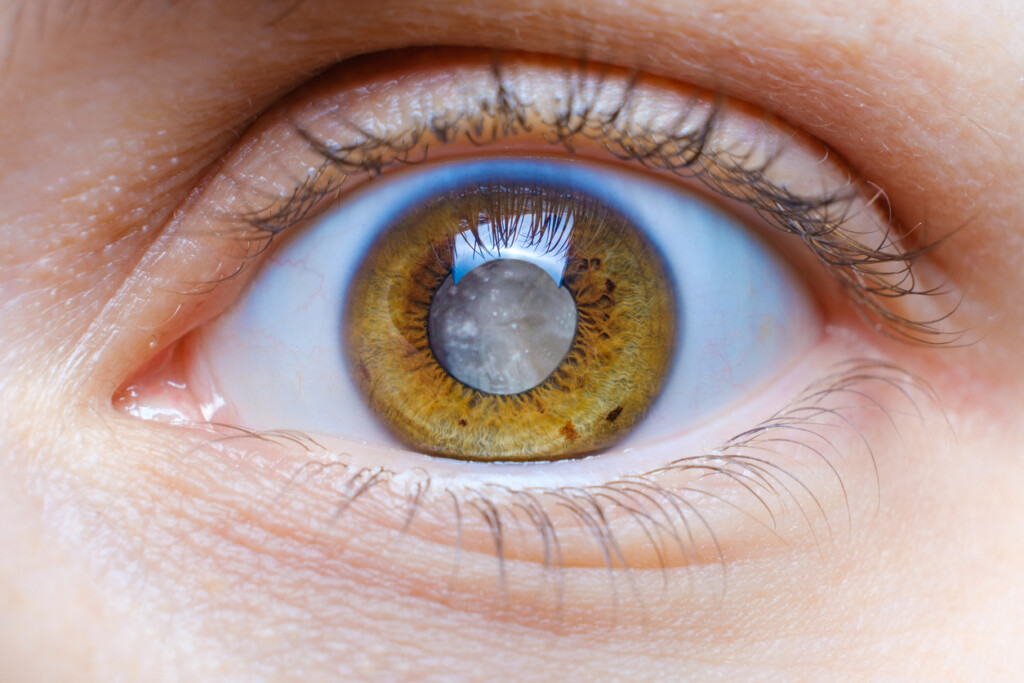
Wie wird ein Retinoblastom diagnostiziert?
Die Diagnose von Retinoblastomen ist mit zahlreichen Untersuchungen verbunden – angefangen von einer umfassenden Überprüfung der Augen und des Augenhintergrunds, die bei Kleinkindern und Säuglingen normalerweise in Narkose durchgeführt wird. Da Retinoblastome sehr selten sind, ist bei der Diagnosestellung Fachkompetenz gefragt, idealerweise erfolgt sie daher in einem medizinischen Zentrum, das sich auf ophthalmologische (Kinder-)Onkologie spezialisiert hat.
Zur Routinediagnostik bei Retinoblastomen gehören unter anderem folgende Untersuchungen
- Augenspiegeluntersuchung und weitere augenärztliche Untersuchungen,
- bildgebende Verfahren wie Ultraschall (Sonografie) und Magnetresonanztomografie (MRT)
- sowie eine genetische Analyse des Blutes, um festzustellen, ob es sich um die erbliche oder nicht-erbliche Variante handelt.

Behandlung von Retinoblastomen
Die jeweils bestmögliche Therapie bei Retinoblastomen ist abhängig von unterschiedlichen Faktoren und setzt eine entsprechende Expertise beim behandelnden Team voraus. Grundsätzlich erfolgt die Behandlung nicht nur jeweils abhängig von der genauen Art des Tumors, sondern auch abhängig davon, wie weit die Erkrankung bereits fortgeschritten ist und ob lediglich eines oder beide Augen betroffen sind. Zudem muss eine realistische Einschätzung der zu erwartenden Sehfähigkeit im Anschluss an die jeweilige Behandlung erfolgen. Und auch das Alter der Patientinnen und Patienten spielt bei der Wahl der optimalen Therapieform eine wichtige Rolle.
Wo können Retinoblastome am besten behandelt werden?
Für die bestmöglichen Heilungschancen von Retinoblastomen sollte die Behandlung in einem speziellen onkologischen Zentrum erfolgen, das verschiedene Fachdisziplinen miteinander vereint und sich auch auf die Therapie dieser seltenen Krebserkrankung spezialisiert hat. Universitätskliniken stellen eine gute Anlaufstelle dar, da hier verschiedene medizinische Fachdisziplinen zusammenkommen. Für die Behandlung von Netzhauttumoren ist die Expertise und Kompetenz unter anderem von Fachleuten aus den Bereichen Augenheilkunde (Ophtalmologie), Pädiatrie, Onkologie, Radioonkologie, Humangenetik und Medizinphysik gefragt.
Retinoblastom-Expertinnen und Experten an der Universitätsmedizin Essen
Die Kinderonkologie der Universitätsmedizin Essen ist eines der deutschlandweit führenden Zentren zur Behandlung von Retinoblastomen. Nehmen Sie Kontakt zu dem Team um Prof Dr. Petra Ketteler, PD Dr. Stefan Schönberger und Susanne Bastian auf. Dort wird die für Ihr Kind passende Therapie interdisziplinär abgestimmt:
Telefon +49 201 – 723 – 2003
Fax +49 201 – 723 – 79002
Mail: RB-Studie@uk-essen.de
Gibt es Leitlinienbehandlungen für Retinoblastome?
Retinoblastome gehören zu den seltenen Krebserkrankungen, was Fachleute generell vor das Problem stellt, dass nicht genügend Daten vorhanden sind, um standardisierte Vorgaben für die Behandlung festzulegen. Bislang wurden zu Retinoblastomen, anders als bei anderen (seltenen) Tumorerkrankungen im Kindes- und Jugendalter, noch keine Therapieoptimierungsstudien durchgeführt. Um Diagnostik und Therapie trotzdem immer weiter zu verbessern und jene Daten zu sichern, die anhand der wenigen jährlichen Fälle existieren, wurde 2013 das Retinoblastomregister eingerichtet. Hier werden die Krankengeschichten von allen Kindern und Jugendlichen unter 18 Jahren aus Deutschland und Österreich dokumentiert, bei denen ein Retinoblastom oder eine Mutation des RB1-Gens festgestellt wurde. Durch die Auswertung der Registerdaten sollen zukünftige Behandlungen weiter verbessert werden. Datensammlung ist Das Register wird von der Kinderonkologie der Universitätsmedizin Essen (Kinderklinik III) geführt.
Podcast PrO-Ton
Die Podcast-Reihe zur Protonentherapie am WPE soll Zuhörenden unser Zentrum und unsere Therapie auf persönlichere Weise näher bringen.

Retinoblastomregister (RB-Register)
Das WPE beteiligt sich im Rahmen seiner umfangreichen Forschungs- und Studientätigkeit auch am Retinoblastomregister, das zentral von der Kinderonkologie der Universitätsmedizin Essen (Kinderklinik III) geleitet wird. Auf diese Weise soll die Protonentherapie für die Behandlung von Patientinnen und Patienten mit Retinoblastomen weiter optimiert werden. Zur bestmöglichen Koordination seiner Forschungstätigkeit hat das WPE ein eigenes Studienbüro etabliert. Weitere Informationen
Wie können Netzhauttumore behandelt werden?
Für die Behandlung eines Retinoblastoms stehen verschiedene Therapieformen zur Verfügung. Eine Operation, bei der das Auge mitsamt Tumor vollständig entfernt wird (Enukleation), ist dabei das sicherste und effektivste Verfahren, insbesondere, wenn der Tumor bereits weit fortgeschritten und das Auge womöglich bereits erblindet ist. Abhängig davon, ob das Retinoblastom bereits gestreut hat, kann der chirurgische Eingriff im Anschluss (adjuvant) mit einer Chemo- oder Strahlentherapie kombiniert werden. Manchmal kommt eine Chemotherapie auch im Vorfeld (neoadjuvant) einer Operation zum Einsatz, um das Retinoblastom zunächst zu verkleinern.
Weitere Therapiemöglichkeiten bei Retinoblastomen zielen darauf ab, das Auge und sein Sehvermögen möglichst zu erhalten. Zu diesen Verfahren gehören:
- Laserkoagulation (Zerstörung des Tumors durch Hitze),
- Kryokoagulation (Vereisung des Tumors),
- Chemotherapie (systemisch oder lokal im Auge),
- Radio-Chemotherapie sowie
- Strahlentherapie.
Die Strahlentherapie als Behandlungsmethode
Retinoblastome gelten als besonders strahlensensibel. Das bedeutet, dass der Tumor besonders gut durch eine Bestrahlung zerstört werden kann. Tatsächlich lässt sich durch eine Strahlentherapie in gut 80 Prozent aller Fälle eine vollständige Tumorkontrolle erreichen – bei gleichzeitigem Erhalt des Auges und eines akzeptablen Sehvermögens.
Allerdings besteht bei einer Strahlentherapie das grundsätzliche Risiko von möglichen Spätfolgen, zum Beispiel der Entwicklung von bösartigen Zweittumoren[AD1] , was insbesondere bei erblichen Retinoblastomen ein Risiko darstellt. Die Vor- und Nachteile einer Bestrahlung müssen daher gerade bei sehr jungen Kindern sorgsam gegeneinander abgewogen werden, auch, um beispielsweise die Gefahr von Entwicklungsstörungen weitmöglichst zu minimieren.
Statt auf eine herkömmliche perkutane Strahlentherapie, die von außen durch die Haut erfolgt, greift man bei Netzhauttumoren daher oft auf eine lokale Bestrahlungsform zurück, die als verträglicher gilt: die Brachytherapie. Dabei wird ein so genannter Strahlenträger in das Auge eingebracht und dort fixiert, bis eine bestimmte Strahlendosis am Tumor abgegeben wurde. Erst dann wird der Strahlenträger wieder entnommen. Beide Eingriffe erfolgen in Vollnarkose.
Als Alternative zur Brachytherapie hat sich in einzelnen Fällen die Protonentherapie etabliert, die als extrem präzise, besonders schonend und nebenwirkungsarm gilt. Eine Bestrahlung mit Protonen ist bei zahlreichen kindlichen Tumoren – vor allem bei jenen in der Nähe von Risikoorganen – bereits fester Standard, zum Beispiel bei Rhabdomyosarkomen oder Ependymomen.
Behandlung von Kleinkindern in Narkose
Bei der Bestrahlung mit Protonen müssen die Patientinnen und Patienten unbedingt vollkommen still liegen. Gerade für Kleinkinder ist das eine große Herausforderung. Für eine erfolgversprechende Protonenbehandlung kann es notwendig und sinnvoll sein, Ihr Kind während der Planung und eigentlichen Therapie zu sedieren. Diese Sedierungen führen am WPE ausschließlich Anästhesisten und Anästhesistinnen durch, die die nötige Erfahrung bei der Behandlung von krebskranken Kindern besitzen. Weitere Informationen zum Thema Anästhesien im WPE haben wir Ihnen auf dieser Seite zusammengestellt.
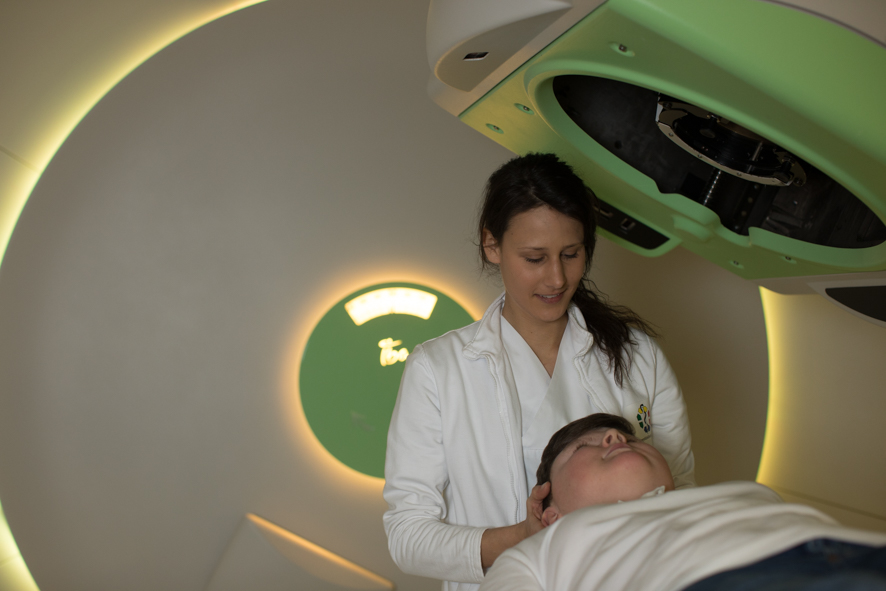
Protonentherapie von Retinoblastomen: Hochmodern, hochpräzise und besonders schonend
Gerade bei Tumoren, die sich in komplizierten Regionen des Körpers entwickeln und/oder in der Nähe von Risikoorganen liegen, hat sich die Protonentherapie mittlerweile als moderne und besonders schonende Bestrahlungsform bewährt. Denn anders als bei der konventionellen Bestrahlung mit Photonen lassen sich Protonen millimetergenau auf den Tumor ausrichten und geben den Hauptteil der Strahlendosis erst unmittelbar im erkrankten Gewebe ab. Das eigentliche Strahlenfeld kann also so klein wie möglich gehalten werden. Bei Augentumoren wie dem Retinoblastom ein nicht zu unterschätzender Vorteil.
Die Protonentherapie wird deshalb auch als hochkonformale Therapie bezeichnet. Konformal bedeutet, dass die Bestrahlung die (unregelmäßigen) Strukturen des Tumors so exakt wie möglich aufgreift und das Bestrahlungsfeld so aufgebaut ist, dass das um den Tumor liegende gesunde Gewebe bestmöglich geschont wird. Das ist gerade bei der Bestrahlung von Tumoren im Kopfbereich besonders wichtig. Denn empfindliche Risikobereiche wie Gehirn, Rückenmark, Seh- und Hörstrukturen werden sehr viel besser geschont als bei einer herkömmlichen Bestrahlung. Zugleich sinkt das grundsätzliche Risiko für potenzielle Nebenwirkungen und Spätfolgen wie beispielsweise Zweittumoren. Bei vielen kindlichen Tumoren hat sich die Protonentherapie daher mittlerweile als Behandlungsstandard etabliert. Schließlich ist gerade bei sehr jungen Kindern das noch unreife Gewebe noch einmal deutlich strahlensensibler als bei erwachsenen Patientinnen und Patienten.
Die Wirksamkeit der Protonentherapie in der Behandlung von Retinoblastomen wurde bereits in Studien belegt. Sie erreicht in der Regel eine sichere Tumorkontrolle – bei gleichzeitigem Erhalt des Auges. Zudem reduziert sie langfristig die Gefahr einer Erblindung. Anders gesagt: Die Bestrahlung mit Protonen gilt mittlerweile als eine der besten konformalen Bestrahlungsmöglichkeiten bei Netzhauttumoren. Und das auch im fortgeschrittenen Stadium der Erkrankung. Tatsächlich ist die Bestrahlung mit Protonen besonders bei großen Tumoren und Blastomen in der Nähe des Sehnervs von Vorteil, denn hier sind die Möglichkeiten der Brachytherapie mit Blick auf die Platzierung und Fixierung von Strahlenträgern begrenzt. Bei anderen Augentumoren, beispielsweise dem Aderhautmelanom, ist die Protonentherapie bereits ein anerkanntes Goldstandard-Verfahren, also die bewährteste und beste Therapieoption, die zur Verfügung steht.

Ihr Kind hat ein Retinoblastom und Sie wollen sich über die Option der Protonentherapie informieren?
Wenn Ihr Kind an einem Retinoblastom erkrankt ist, ist als erste Anlaufstelle eine auf Retinoblastome spezialisierte Klinik – wie die Kinderonkologie der Universitätsmedizin Essen – zu kontaktieren. Bei Fragen zur Protonentherapie beim Retinoblastom nehmen Sie gerne zu unserem Case-Management Kontakt auf.
Case Management: 0201 723 6600 oder wpe@uk-essen.de
Kann die Protonentherapie mit anderen Behandlungsmethoden kombiniert werden?
Die Protonentherapie kann ebenso wie eine konventionelle Bestrahlung mit Photonen mit anderem Therapiemöglichkeiten kombiniert werden – beispielsweise mit einer Chemotherapie und selbstverständlich auch mit einer Operation.
Welche Nebenwirkungen hat die Protonentherapie von Retinoblastomen?
Eine Bestrahlung ist mit Nebenwirkungen verbunden, ganz gleich, ob konventionell mit Photonen oder mit Protonen bestrahlt wird. Bei der Protonentherapie handelt es sich jedoch um eine lokale Therapiemaßnahme, die ihre Wirkung nicht im gesamten Körper, sondern nur in der vorab definierten Region um den Tumor entfaltet. Zudem fallen die Nebenwirkungen aufgrund der Zielgenauigkeit, mit der die Tumorregion bestrahlt wird, deutlich geringer aus als bei einer herkömmlichen Strahlentherapie.
Bei der Bestrahlung von Retinoblastomen mit Protonen sind unter anderem folgende Nebenwirkungen möglich:
- Linsentrübung (Grauer Star),
- erhöhter Augeninnendruck,
- Gefäßveränderungen sowie
- Augentrockenheit.
- Sehminderung
- Assymetrisches Knochenwachstum der Orbita
Diese und je nach Bestrahlungsgebiet weitere mögliche Spätfolgen treten aufgrund der punktgenauen Bestrahlung mit Protonen jedoch nur selten auf.
Protonenbestrahlung am WPE der Universitätsmedizin Essen
Das WPE (Westdeutsches Protonentherapiezentrum Essen) ist eines der führenden Zentren für Protonenbestrahlung in Deutschland. Es ist Teil der Universitätsmedizin Essen und an das Westdeutsche Tumorzentrum Essen (WTZ) angebunden. Das WTZ ist das älteste und zugleich eines der größten Tumorzentren Deutschlands sowie eines von nur 13 anerkannten deutschen Spitzenzentren für Tumorforschung, -diagnostik und -therapie nach dem US-amerikanischen Vorbild eines Comprehensive Cancer Centers (CCC). Es verfügt über eine OnkoZert-Zertifizierung und ist in das Deutsche Konsortium für Translationale Krebsforschung (DKTK) eingebunden. Das WPE hat sich auf die Behandlung von Kindern und Jugendlichen spezialisiert. Weitere Informationen
Welche Vorteile hat eine Protonentherapie am WPE?
Das WPE ist eines der fortschrittlichsten Protonentherapiezentren der Welt. Grundsätzlich behandeln wir eine Vielzahl an Tumoren, die aufgrund ihrer Lage oder des jungen Alters der Patientinnen und Patienten eine möglichst genaue Bestrahlung erfordern. Dies geschieht in fachübergreifender Zusammenarbeit und über die Teilnahme an interdisziplinären Tumorkonferenzen mit den Kliniken der Universitätsmedizin Essen, darunter unter anderem mit der Klinik für Augenheilkunde und der Kinderonkologie der Universitätsmedizin Essen (Kinderklinik III).

Eigener Therapieplatz zur Behandlung bösartiger Augentumoren
In Kooperation mit der Klinik für Augenheilkunde der Universitätsmedizin Essen hat die Klinik für Partikeltherapie am WPE 2021 einen speziellen Augentherapieplatz für die Bestrahlung maligner Augentumore in Betrieb genommen. An der so genannten Eyeline sitzen Patientinnen und Patienten an einem eigens angefertigten Stuhl vor dem Gerät, aus dem der Protonenstrahl kommt. Auf diese Weise kann das Tumorgewebe immer absolut exakt getroffen werden, egal in welchem Bereich der Tumor auftritt. Weltweit ist die Universitätsmedizin Essen damit das einzige Tumorzentrum , das über alle Behandlungsmodalitäten zur Bekämpfung von uvealen Melanomen an einem Standort verfügt. Mehr zu Behandlung von Aderhautmelanomen am WPE
Das WPE stellt das größte Bestrahlungsprogramm für Kinder in Europa
Das WPE steht für Kompetenz und Erfahrung vor allem in der pädiatrischen Onkologie: Die Protonenbestrahlung von Patientinnen und Patienten im Kindes- und Jugendalter ist einer der ausgewiesenen Schwerpunkte des WPE. Durch Zuweisungen aus ganz Deutschland, Europa und darüber hinaus behandeln wir jährlich bis zu ca. 300 Kinder und Jugendliche im Alter von bis zu 18 Jahren, über die Hälfte unserer jungen Patientinnen und Patienten ist unter sieben Jahre alt ist. Gemessen an der relativ geringen Häufigkeit kindlicher Tumore ist das im europäischen Vergleich ein hoher Anteil.
Tatsächlich stellt das WPE das größte Bestrahlungsprogramm für Kinder in Europa. Da Tumoren bei Kindern zudem in vielfältiger Form in Erscheinung treten, haben wir über die Jahre hinweg eine fundierte Fachkompetenz bei zahlreichen kindlichen Krebserkrankungen (Indikationen) aufgebaut. Weitere Informationen zur Behandlung von Kindern und Jugendlichen am WPE haben wir Ihnen auf dieser Seite zusammengestellt.
Pencil Beam Scanning – die fortschrittlichste Art der Protonentherapie
Das WPE nutzt die fortschrittlichste Art der Protonentherapie – das Pencil Beam Scanning. Bei dieser Technik bestrahlen wir ein Retinoblastom mit einem bleistiftspitzendünnen Strahl, den wir Punkt für Punkt über den gesamten dreidimensionalen Raum des Tumors steuern. Auf diese Weise bestrahlen wir nahezu ausschließlich das erkrankte Gewebe und schonen die umliegenden Regionen bestmöglich.
Psychosoziale Versorgung
Das psychosoziale Team des WPE unterstützt Sie und Ihre Familie während der Zeit der Strahlentherapie optimal. Das Team übt gemeinsam mit Ihrem Kind spielerisch die Elemente der Behandlungssituation ein und etabliert zudem kleine Belohnungsrituale während der eigentlichen Therapie. So nehmen wir Ihnen und Ihrem Kind mögliche Ängste und machen Ihrem Kind die Behandlung am WPE so angenehm und einfach wie irgend möglich. Mehr zu diesem Konzept erfahren Sie auf dieser Seite.
Wer kann mit der Protonentherapie am WPE behandelt werden?
Am WPE bieten wir Patientinnen und Patienten aus Deutschland und dem Ausland die Möglichkeit einer nebenwirkungsarmen Tumortherapie an. Dies geschieht in Zusammenarbeit mit den Kliniken der Universitätsmedizin Essen und dem Westdeutschen Tumorzentrum (WTZ), also mit allen für die Behandlung notwendigen Fachdisziplinen. Von dieser gebündelten onkologischen Expertise profitieren alle Patientinnen und Patienten des WPE, insbesondere aber jene, deren Heimatklinik weit entfernt ist und die onkologische Kompetenz vor Ort benötigen. So ist beispielsweise eine parallele Chemotherapie am Universitätsklinikum Essen möglich. Grundsätzlich kann diese jedoch auch am Heimatkrankenhaus erfolgen.
Protonenbestrahlung von Patientinnen und Patienten aus dem Ausland
Als erfahrenes Zentrum für Protonentherapie unterstützen wir Sie natürlich auch, wenn Sie im Ausland leben. Wir haben große Erfahrung mit internationalen Patientinnen und Patienten und können bei vielen Ländern auf bereits bestehende strukturierte Kooperationsprogramme zurückgreifen. Daher können wir Sie von der anfänglichen Organisation bis zur Therapie und darüber hinaus vollumfänglich begleiten
Wie sieht der Behandlungsablauf am WPE aus?
Sollten Sie sich für eine Bestrahlung des Retinoblastoms Ihres Kindes mit Protonen am WPE interessieren, stellen Sie oder Ihr behandelnder Arzt bzw. Ihre Ärztin zuallererst eine Therapieanfrage an uns.
Unser Case-Management-Team nimmt die Anfrage dann entgegen und steht von da an für Sie als Ansprechperson zur Verfügung. Fragen, die vom Case-Management nicht beantwortet werden können, leitet dieses umgehend an die behandelnden Ärzte und Ärztinnen weiter.
Nach der Therapieanfrage ist der Ablauf wie folgt:
Wenn uns alle relevanten Informationen und Dokumente vorliegen, entscheidet das zuständige ärztliche Team, ob eine Protonenbestrahlung in Ihrem Fall sinnvoll ist. Ihre individuelle Anfrage wird zudem mit den Kolleginnen und Kollegen aus anderen relevanten Fachdisziplinen der Universitätsmedizin Essen und im interdisziplinären Onkologie-Tumorboard besprochen – einer Gesprächsrunde aus fachlich versierten Expertinnen und Experten. Gegebenenfalls werden Ihnen im Anschluss auch alternative Behandlungsoptionen empfohlen.
Sollte sich das Expertenteam zur Behandlung des Retinoblastoms mit der Protonentherapie entscheiden, beginnt die Therapieplanung. Hierfür laden wir Sie und wenn gewünscht auch einen Angehörigen oder eine Angehörige zu einem Termin ein. In diesem erläutern wir Ihnen ausführlich die Chancen und Risiken der Protonenbestrahlung Retinoblastomen.
Nach Ihrem Einverständnis zur Protonenbestrahlung beginnen die Vorbereitungen der Therapie, daran anschließend startet die Protonentherapie mit folgenden Schritten:
- Anfertigung einer individuellen Lagerungshilfe, damit das Retinoblastom Ihres Kindes in jeder Bestrahlungssitzung exakt mit Protonen bestrahlt werden kann.
- Nutzung bildgebender Verfahren, wie beispielsweise CT und gegebenenfalls auch MRT, damit unser ärztliches Team und die Medizinphysiker und Medizinphysikerinnen des WPE die genaue Lage des Tumors und der umliegenden Organe beurteilen können.
Nach der Planung und Qualitätssicherung beginnt ein bis zwei Wochen später schließlich die tägliche Protonentherapie. Die Sitzungen finden ambulant statt und gehen im Rahmen der Studien über einen Zeitraum von etwa vier Wochen. Die einzelne Strahlenbehandlung dauert dabei meist nicht länger als eine halbe Stunde, wobei die eigentliche Protonenbestrahlung sogar nur wenige Minuten in Anspruch nimmt.
Welche Informationen sind für die Erstvorstellung nötig?
Sollten Sie sich für eine Protonenbestrahlung eines Retinoblastoms interessieren, benötigen wir für die Prüfung folgende Dokumente:
- den Histologiebefund,
- radiologische Befunde,
- zusammenfassender Arztbericht,
- OP-Berichte,
- aktuelle CT- und MRT-Bilder,
- Pathologiebefunde.
Diese Unterlagen können entweder Sie selbst einreichen oder Ihr behandelnder Arzt oder Ihre Ärztin. Auf Basis der vorliegenden Dokumente entscheiden unsere Radioonkologinnen und -onkologen dann, ob eine Protonentherapie bei Ihrem Kind möglich ist. Sollte dies der Fall sein, benötigen wir zu einem späteren Zeitpunkt wahrscheinlich weitere Dokumente. Gegebenenfalls fordern wir auch weitere Untersuchungen an, um eine optimale Therapieplanung durchzuführen.
Unser Case-Management informiert Sie oder Ihren behandelnden Arzt/Ihre behandelnde Ärztin in jedem Fall rechtzeitig darüber, so dass die Therapie zeitnah beginnen kann.
Kostenübernahme der Protonentherapie
Mit diversen gesetzlichen Krankenkassen haben wir Verträge zur Übernahme der Kosten einer Protonentherapie von Retinoblastomen geschlossen. Mit anderen und auch mit einigen privaten Krankenkassen haben wir Abläufe zur Kostenübernahme etabliert. Wir unterstützen alle Patientinnen und Patienten bei der Kostenklärung direkt von Beginn an.
Kontaktaufnahme
Sie möchten sich über die Protonentherapie eines Retinoblastoms am WPE informieren? Oder direkt einen Termin machen? Dann nutzen Sie unsere nachfolgenden Kontaktmöglichkeiten.
Jede neue Kontaktaufnahme erfolgt über unser erfahrenes und engagiertes Case-Management – der Schnittstelle zwischen Ihnen und unserem Team. Es unterstützt Sie bei
- der Zusammenstellung Ihrer Unterlagen,
- bei der Kostenübernahme,
- bei Reise und Unterkunft
und beantwortet alle Ihre aufkommenden Fragen. Es stellt auch den Kontakt zu unseren Strahlentherapeuten und -therapeutinnen her.
Case-Management
E-Mail: wpe@uk-essen.de
Tel: 0201 723 6600
Selbsthilfeorganisation:
Die KAKS hilft betroffenen Kindern und Erwachsenen und ihren Familien.
Forschung zum RB am WPE:
Im WPE erarbeitet mit der Förderung der „Ernst und Berta Grimmke Stiftung“ ein Forschungsprojekt Strategien zur Minimierung des Risikos sekundärer Malignome nach einer Protonentherapie bei der erblichen Form des Retinoblastoms.
Kinderkrebsinfo
Die Kinderkrebsinfo hat auch Informationen zur Behandlung von Retinoblastomen zusammengestellt.